
Enfermedades Genéticas II
Paquioniquia Congénita

La
base genética de la paquioniquia congénita. En 1994 se dilucidaron las bases
moleculares de la paquioniquia congénita (PC). Cuatro genes de queratina están
asociados con los principales subtipos de PC: Los defectos en K6a o K16 causan
la PC-1; y las mutaciones en K6b o K17 causan la PC-2. Las mutaciones en las
queratinas, las proteínas de filamentos intermedios específicas del epitelio,
dan lugar a redes citoesqueléticas aberrantes que se presentan clínicamente
como una variedad de fenotipos de fragilidad epitelial. Hasta la fecha, las
mutaciones en 20 genes de las queratinas están asociadas a trastornos humanos.
Aquí, revisamos las bases genéticas de la PC y reportamos 30 nuevas mutaciones
de la PC. De ellas, 25 mutaciones se encontraron en familias de PC-1 y cinco
mutaciones se identificaron en familias de PC-2. Todas las mutaciones
identificadas mutaciones de aminoácidos heterocigotas o pequeñas mutaciones de
deleción dentro del marco, con la excepción de una mutación inusual en un caso
esporádico de PC-1. Este último portaba una duplicación de 117 pb que daba
lugar a una inserción de 39 aminoácidos en el dominio 2B de K6a.
También
cabe destacar la mutación L388P en K17, que es el primer defecto genético
identificado en el motivo de terminación de la hélice de esta proteína. La
comprensión de las bases genéticas de estos trastornos permite un mejor
asesoramiento para los pacientes y allana el camino para el desarrollo de
terapias.
La
paquioniquia congénita (PC) es una forma rara de queratodermia palmoplantar
hereditaria (PPK), de aparición extremadamente infrecuente. En la literatura dermatológica,
la PC es más conocida como síndrome de Jadassohn-Lewandowsky. La enfermedad es
rara, pero se han descrito más de 250 casos. Se han propuesto varias
clasificaciones para la PC. Actualmente se reconocen dos síndromes distintos de
PC:
PC-1 (el tipo Jadassohn-Lewandowsky)
PC-2 (tipo Jackson-Lawler).
Etiología.
La PC es el resultado de mutaciones en los genes que codifican las queratinas
de los queratinocitos epidérmicos, concretamente K6a, K6b, K16 y K17. En la
mayoría de los casos, se describe un modo de herencia autosómico dominante; sin
embargo, en la literatura también se menciona la herencia autosómica recesiva.
Cockayne (1933) fue el primero en expresar la opinión de que la presencia de un
factor adicional, probablemente una
segunda mutación genética, es necesaria para la expresión de la enfermedad.
Munro (1994) fue el primero en proponer que el defecto genético de la PC está
vinculado al grupo de genes de la queratina en el cromosoma 17.
Características clínicas. Las lesiones cutáneas de la paquioniquia congénita suelen aparecer poco después del nacimiento; ambos sexos se ven afectados por igual y consisten en cambios distróficos en las uñas de las manos y los pies, callos hiperqueratósicos en las palmas de las manos y las plantas de los pies, queratosis folicular alrededor de las rodillas y los codos, e hiperhidrosis o sudoración excesiva en las manos y los pies. Los cambios en las uñas, de los que deriva el nombre de la enfermedad, consisten en un marcado engrosamiento, que aumenta hacia el borde libre, y el lecho ungueal se llena de restos queratósicos amarillentos, lo que a menudo hace que la uña se proyecte hacia arriba en el borde libre. También se han descrito casos de pelo escaso y disqueratosis corneal que producen opacidades en la córnea.
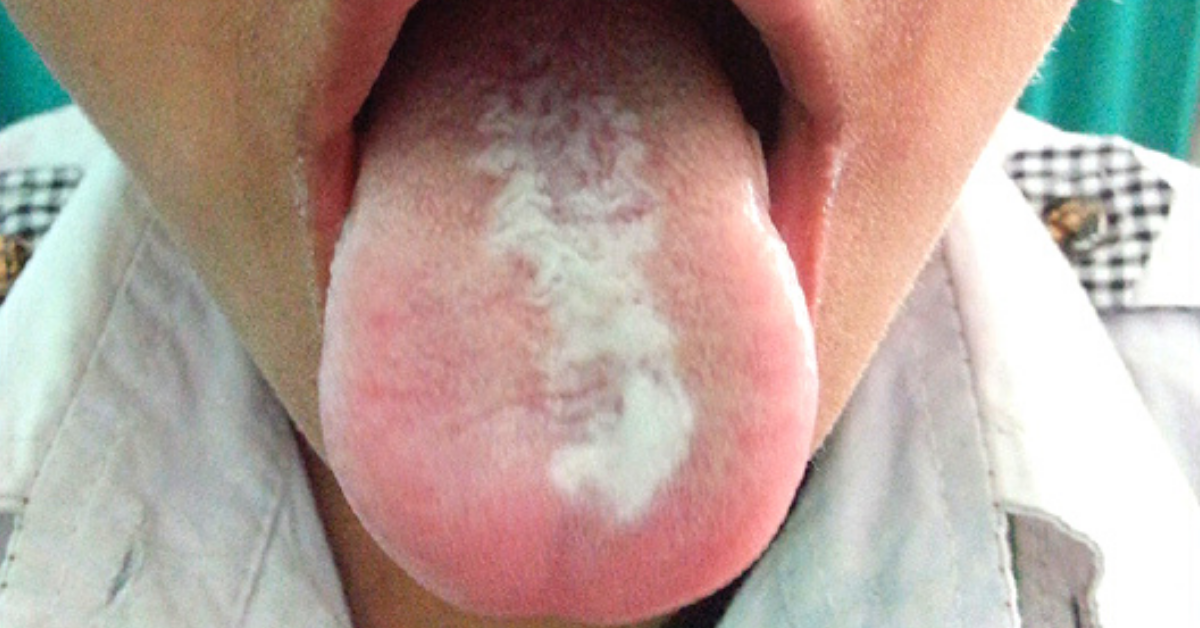
Manifestaciones orales.
Según Gorlin y Chaudhry, las lesiones orales están casi siempre presentes.
Consisten en un engrosamiento focal o generalizado, blanco y opaco, de la
mucosa que afecta a la mucosa bucal, la lengua o los labios. Estas lesiones
orales leucoplásicas no deben confundirse clínicamente con el liquen plano. Se
ha informado de que incluso están presentes al nacer. También se ha informado
de la presencia de queilosis angular. En varias ocasiones se han encontrado
dientes presentes en el nacimiento, dientes natales. Maser y Young y Lenox han
informado de casos con lesiones orales típicas.

Hallazgos histológicos.
Las lesiones hiperqueratósicas de la piel y la mucosa oral muestran acantosis,
hiperqueratosis y paraqueratosis.
No
se observan cambios premalignos. La microscopía electrónica muestra filamentos
intermedios engrosados y agrupados, así como gránulos de queratohialina
ampliados. En la capa granular ensanchada se observan masas gruesas de
tonofilamentos y gránulos de queratohialina grandes e irregulares. En la capa
espinosa, se encuentran masas gruesas de tonofilamentos en la periferia de las
células.
El
diagnóstico diferencial debe incluir la leucoplasia, el liquen plano, el nevo
blanco esponjoso, la disqueratosis congénita, la disqueratosis intraepitelial
benigna hereditaria y el síndrome de hiperqueratosis focal palmoplantar y de la
mucosa oral.
Tratamiento.
En la actualidad, no existe ningún tratamiento para esta enfermedad, que no se
considera grave.
Disqueratosis
Congénita

La
disqueratosis congénita (DKC) es una genodermatosis bien reconocida pero rara,
caracterizada por hiperpigmentación cutánea reticulada, distrofia ungueal,
leucoplasia premaligna de la mucosa oral y pancitopenia progresiva. La
importancia del síndrome radica en la alta incidencia de cáncer oral que se
desarrolla en los adultos jóvenes afectados.
Etiología.
Se ha demostrado que las mutaciones en el DKC1 causan la forma ligada al
cromosoma X del DKC. El patrón de herencia de la mayoría de los casos de DKC es
recesivo ligado al cromosoma X, pero se han descrito patrones autosómicos
dominantes y recesivos.
Características
clínicas. La disqueratosis congénita es un síndrome poco frecuente, con
aproximadamente 180 individuos descritos en la literatura. Dado que este
trastorno es principalmente recesivo ligado al cromosoma X, la proporción entre
hombres y mujeres es de aproximadamente 10:1. Los cambios en las uñas suelen
ser la primera manifestación de la enfermedad, volviéndose distróficas y
desprendiéndose algún tiempo después de la edad de cinco años. La pigmentación
marrón-grisácea de la piel aparece al mismo tiempo o unos años después y se
distribuye por el tronco, el cuello y los muslos. La piel puede volverse
atrófica y telangiectásica y la cara aparece enrojecida.
También
se han descrito casos ocasionales con un amplio espectro de otras
manifestaciones menores que incluyen un esqueleto frágil, retraso mental, silla
turca pequeña, disfagia, membranas timpánicas transparentes, sordera, epífora e
infecciones de los párpados, anomalías uretrales, testículos pequeños,
anomalías dentales y, comúnmente, hiperhidrosis de la palma de la mano y las
plantas de los pies.
Manifestaciones
orales. La leucoplasia de la mucosa suele aparecer en la mucosa bucal y puede
afectar a la lengua y la orofaringe. La leucoplasia puede volverse verrugosa y
puede producirse una ulceración.
Pueden
verse afectadas otras zonas de la mucosa (por ejemplo, el esófago, el meato
uretral, el glande del pene, el conducto lagrimal, la conjuntiva, la vagina y
el ano). Puede producirse constricción y estenosis, con el desarrollo de
disfagia, disuria, fimosis y epífora. Los pacientes tienen una mayor incidencia
de neoplasias malignas, en particular carcinoma de células escamosas de la
piel, la boca, la nasofaringe, el esófago, el recto, la vagina y el cuello
uterino. Éstos suelen producirse dentro de las zonas de leucoplasia. Los
pacientes también pueden tener una mayor incidencia y gravedad de caries
dentales y pérdida de dientes.

Hallazgos
histológicos. Las muestras de biopsia de piel de las zonas de pigmentación
reticulada suelen mostrar una hiperqueratosis leve, atrofia epidérmica,
telangiectasia de los vasos sanguíneos superficiales y melanófagos en la dermis
papilar. También se han descrito cambios en la interfase, con una leve
vacuolización de la capa basal y un infiltrado inflamatorio linfocítico en la
dermis superior. Las lesiones orales no se han estudiado a fondo, pero las
lesiones leucoplásicas parecen ser hiperparqueratosis o hipercoqueratosis y
acantosis inespecíficas. Según el estadio de la enfermedad, el epitelio puede
mostrar displasia. No se ha descrito la naturaleza exacta de las vesículas y
úlceras precedentes.
Hallazgos
de laboratorio. Muchos casos se han caracterizado también por cambios
hematológicos que incluyen anemia, leucopenia, trombocitopenia y pancitopenia.
Algunos pacientes han desarrollado anemia de Fanconi. De hecho, se ha sugerido
que el síndrome de Fanconi, o la pancitopenia familiar de Fanconi, es
simplemente una expresión variada de la disqueratosis congénita.
El
diagnóstico diferencial de las lesiones orales debe incluir la leucoplasia, el
liquen plano, la paquioniquia congénita y la epidermólisis bullosa.
Las
pruebas de laboratorio útiles para el diagnóstico son el examen de las
células sanguíneas y los niveles bajos de gammaglobulina sérica.
Tratamiento.
Las opciones de tratamiento a corto plazo para la insuficiencia de la médula
ósea en pacientes con DKC incluyen la eritropoyetina y el factor estimulante de
colonias de granulocitos; sin embargo, la única opción curativa a largo plazo
es la transferencia de médula ósea alogénica. La alta frecuencia de
transformación maligna de las lesiones orales requeriría un examen periódico
cuidadoso del paciente para detectar tal ocurrencia.
Displasia Ectodérmica Hipohidrótica
La
displasia ectodérmica hipohidrótica se caracteriza por cambios displásicos de
los tejidos de origen ectodérmico y suele heredarse como un rasgo recesivo
ligado al cromosoma X.
recesivo
ligado al cromosoma X, por lo que afecta principalmente a los varones.
Características clínicas.
Los
recién nacidos con HED pueden presentar una piel descamada similar a la de los
bebés "post-maduros".
La
función ecrina (sudoración), aunque está presente, es muy deficiente, lo que
provoca episodios de hipertermia. Lo más frecuente es que el diagnóstico se
retrase hasta que los dientes no erupcionan a la edad esperada (6-9 meses) o
los dientes que erupcionan tienen forma de clavija, cónica o de cuchillo, lo
que puede afectar a la capacidad de comer y hablar. Los pacientes también
tienen una facies peculiar, caracterizada por hiperpigmentación periorbital,
puente nasal deprimido (deformidad de la nariz en forma de silla de montar),
mentón puntiagudo, saliente frontal, labios evertidos, hipoplasia del tercio
medio facial. Suelen tener el cuero cabelludo y el vello corporal escasos
(hipotricosis), a menudo de color claro y de crecimiento lento; las cejas y las
pestañas son escasas o están totalmente ausentes. Las anomalías en la función
de la membrana mucosa provocan frecuentes infecciones de las vías respiratorias
y cambios en las secreciones nasales, desde concreciones (secreciones
solidificadas en las fosas nasales y auditivas) en la primera infancia hasta
grandes coágulos mucosos. La epidermis es xerótica, con parches de
hiperqueratosis y/o eczematosa.
Las
manifestaciones otorrinolaringológicas comunes incluyen infecciones crónicas
como rinitis, faringitis, otitis media, pérdida de audición, epistaxis y
disfonía. Como consecuencia de la hipoplasia de las glándulas gastroentéricas,
los pacientes con DEH también pueden sufrir disfagia y estreñimiento. Por lo
demás, el crecimiento físico y el desarrollo psicomotor están dentro de los
límites normales. En el HED están afectados los varones, pero las mujeres
portadoras pueden manifestar rasgos más leves: agenesia dental congénita y
dientes deformes, pelo escaso y fino y algunos problemas en la función de las
glándulas sudoríparas.
La
Fundación Nacional para las Displasias Ectodérmicas (NFED) ha creado una base
de datos de pacientes que ha permitido determinar las características clínicas
más frecuentes en un amplio grupo de pacientes, en base a lo cual, la
característica más reportada en los pacientes con HED es la hipohidrosis, seguida
de la hipotricosis y la hipodoncia igualmente representadas entre los
pacientes. Otras complicaciones descritas principalmente en pacientes
masculinos son la congestión nasal con mal olor que interfiere con la
alimentación, el eczema y la sinusitis recurrente.
Se
han descrito varias veces diferencias de fenotipo inter e intrafamiliar en los
pacientes, presentando un amplio espectro de gravedad sin una correlación
genotipo-fenotipo. Sin embargo, se han descrito pocas mutaciones hipomórficas
en las que se haya establecido una correlación genotipo-fenotipo para la
función de las glándulas sudoríparas o los signos de la piel y el cabello.
Características
dentales

Las
anomalías dentales de los pacientes con HED incluyen agenesias dentales que van
desde la hipodoncia (pacientes a los que les faltan hasta 5 dientes) hasta la
oligodoncia (pacientes a los que les faltan más de 5 dientes), las
malformaciones dentales son igualmente frecuentes como la microdoncia, los
dientes en forma de cono y la erupción dental anómala.
En
los pacientes con HED también se encuentra el taurodontismo, que se caracteriza
por el agrandamiento de la pulpa. Además, el grado de taurodontismo parece
estar asociado al gen mutado, por lo que se ha propuesto que podría ser más
grave cuando el gen EDA está mutado. Todas estas características podrían dar
lugar a otras anomalías como la reabsorción del hueso alveolar, la osteopenia y
la reducción de la altura.
Las
malformaciones dentales pueden afectar a la función oral y a la autoestima de
los pacientes, por lo que la rehabilitación dental en edades tempranas suele
ser elegida por el equipo de odontólogos especialistas, siendo la terapia con
implantes la técnica clínica más utilizada en estos pacientes.
La
rehabilitación oral debe ser un trabajo de equipo multidisciplinar, que incluya
al odontopediatra, al ortodoncista, al endodoncista, al cirujano maxilofacial y
al prostodoncista para planificar el mejor manejo del paciente. Aunque pueden
surgir riesgos potenciales, el uso de implantes parece ser la mejor opción para
estos pacientes, por lo que deben tenerse en cuenta el mejor momento y lugar
para la colocación del implante y las posibles consecuencias con respecto a la
erupción normal de los dientes y el crecimiento normal de la dentadura.
Diagnóstico
molecular y tratamiento futuro
Hasta
la fecha hemos diagnosticado las mutaciones de EDA con la secuenciación de
Sanger, aunque se han comunicado pocos estudios sobre NGS (Next Generation
Sequencing) que detecten la mutación de EDA.
Dado
que el coste de la secuenciación del exoma está disminuyendo rápidamente, la
aplicación de este método, que ahora es rentable y más barato que el enfoque
clásico del análisis de ligamiento seguido de la secuenciación Sanger de los
genes candidatos del locus identificado, podría considerarse en el futuro como
una herramienta clínica-molecular de diagnóstico.
La
propuesta de nuevas estrategias diagnósticas puede permitir un diagnóstico más
rápido dando la posibilidad de establecer patrones de herencia útiles para las
familias, y de responder a los científicos "sedientos".
Hasta la fecha, sólo se dispone de tratamiento sintomático para estos pacientes. Se espera que los enfoques terapéuticos causales de estos trastornos sean eficaces en un futuro próximo.
Los
rasgos clínicos son una facies característica con protuberancia frontal, labios
y orejas grandes y nariz en forma de silla de montar; piel fina y seca y pelo
corto rubio y escaso, disminución de la sudoración o anhidrosis completa,
debido a la ausencia de glándulas sudoríparas; ausencia de cejas; y lesiones
orales.
El
hallazgo característico en la cavidad oral es la hipodoncia o la anodoncia.
Cuando los dientes están presentes, son hipoplásicos y suelen tener una forma
cónica. En algunos casos puede producirse xerostomía como resultado de la
hipoplasia de las glándulas salivales. La enfermedad suele presentarse durante
el primer año de vida, con fiebre de causa desconocida junto con el retraso en
la erupción o la ausencia de los dientes deciduos.
El
diagnóstico diferencial incluye la oligodoncia idiopática, el síndrome de
Papillon-Lefevre, la displasia condroectodérmica, la displasia cleidocraneal y
la hipoplasia dérmica focal.
Las
pruebas de laboratorio útiles para establecer el diagnóstico son las
radiografías dentales y la demostración de hipohidrosis o anhidrosis.
Tratamiento. No existe un tratamiento específico. Sin embargo, se deben construir prótesis parciales o completas lo antes posible.
Conclusiones
Los
DEH se han asociado a mutaciones en diferentes genes, sin que exista una
correlación genotipo-fenotipo bien establecida, lo que dificulta el diagnóstico
y el establecimiento del patrón de herencia sin la secuenciación del ADN.
Por
lo tanto, es necesario un grupo de especialistas multidisciplinares que incluyan
al odontopediatra, al dermatólogo, al oftalmólogo, al genetista y al
otorrinolaringólogo para un buen manejo del paciente.
La secuenciación del exoma puede ser el mejor enfoque disponible para el diagnóstico molecular, reduciendo el tiempo y el coste y aumentando la sensibilidad del diagnóstico. La rehabilitación oral temprana es obligatoria para mejorar la calidad de vida.



Comentarios
Publicar un comentario